All published articles of this journal are available on ScienceDirect.

Bilateral Temporal Bone Langerhans Cell Histiocytosis: Radiologic Pearls
Authors Info & Affiliations
Abstract
Langerhans cell histiocytosis (LCH) is a rare histiocytic disorder with an unpredictable clinical course and highly varied clinical presentation ranging from single system to multisystem involvement. Although head and neck involvement is common in LCH, isolated bilateral temporal bone involvement is exceedingly rare. Furthermore, LCH is commonly misinterpreted as mastoiditis, otitis media and otitis externa, delaying diagnosis and appropriate therapeutic management. To improve detection and time to treatment, it is imperative to have LCH in the differential diagnosis for unusual presentations of the aforementioned infectious head and neck etiologies. Any lytic lesion of the temporal bone identified by radiology should raise suspicion for LCH. We hereby describe the radiologic findings of a case of bilateral temporal bone LCH, originally misdiagnosed as mastoiditis.
INTRODUCTION
Langerhans cell histiocytosis (LCH) refers to a rare group of diseases most commonly characterized by abnormal clonal proliferation of Langerhans cells in one or more systems, most commonly bone. Previously identified diseases such as histiocytosis-X, Letterer-Siwe disease, Hand-Schuller-Christian disease, diffuse reticuloendotheliosis, and eosinophilic granuloma are now categorized as “LCH” [1].
LCH can be diagnosed in any age group, but it most commonly affects children between 1 and 4 years of age with an incidence of 5-6 cases per million children [1-3]. Head and neck involvement, primarily skull base, is noted in about 60% of the cases. Temporal bone involvement occurs in 15-25% of LCH cases with bilateral disease in 25-30% of those cases [1, 4, 5]. The clinical course of LCH is unpredictable and can range from spontaneous resolution to rapidly progressive and even fatal disease. Radiographic studies are considered to be the most accurate investigation for the detection of bony LCH.
The prognosis of localized temporal bone LCH in children is good, with a survival rate of over 90%. In multifocal disease, the survival rate is 65-100% [1, 2, 6]. Prognostic factors include age at presentation, multisystem involvement, and end organ dysfunction. Children less than 2 years old have a worse outcome [1, 3]. The emphasis of this article is to identify radiologic clues to diagnose temporal bone LCH early in its course, and thus initiate appropriate therapy promptly.
CASE REPORT
A previously healthy 2-year-old girl presented with a ten day history of progressive left-sided postauricular erythema and swelling. She was noted to have clinical worsening while being treated with antibiotics, and was referred to our hospital for further evaluation for acute mastoiditis.
On examination, the patient's left ear was protuberant anteriorly from swelling posteriorly. There was overlying erythema and swelling in the postauricular area. This area was very soft, and a bony edge was palpated surrounding the soft area. It was not warm or tender. External auditory canal examination showed the posterior canal wall to be pushed forward, impacting the anterior canal wall. The posterior canal wall had a pink, irregular surface consistent with granulation-like changes. The right ear exam was normal. No skin lesions or ear drainage were noted. Neurologic exam was normal. The remainder of the exam was unremarkable.
Laboratory studies revealed a mild normocytic anemia (9.8 g/dL; reference range 11.0 to 14.0 g/dL), a mild leukocytosis (11.2 x10(9)/L; reference range 6.0 to 11.0 x10(9)/L), an elevated C-reactive protein (58 mg/L; reference range < 8 mg/L) and erythrocyte sedimentation rate (64 mm/hour; reference range <22 mm/hour).
The skull radiograph demonstrated lytic lesions of the bilateral temporal bones (Fig. 1). Imaging with computed tomography (CT) revealed bilateral bony erosion of the temporal bone in the petromastoid region, left greater than right, with left-sided extension into the soft tissue (Fig. 2). Differential diagnosis based on imaging included unusual otomastoiditis, Langerhans cell histiocytosis, rhabdomyosarcoma, lymphoma, metastasis such as neuroblastoma, or unusual Wegener’s granulomatosis. For further characterization of these lesions and to evaluate for the presence of intracranial extension, magnetic resonance imaging (MRI) was conducted (Fig. 3). Again, bilateral temporal bone changes and left sided soft tissue prominence were noted. This extended superiorly, along the tegmen tympani where there was minimal adjacent dural thickening and enhancement without brain parenchymal involvement (Fig. 3d). These lesions enhanced with gadolinium. Given the sparing of the labyrinth (Fig. 2b and 3b), the bilaterality, and the overall appearance, the findings were most consistent with LCH.
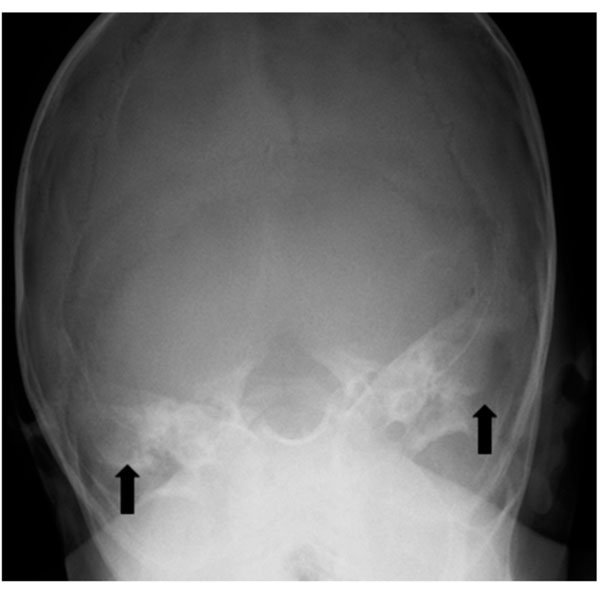
Frontal radiograph of the skull demonstrates lytic changes of the temporal bones bilaterally (arrows).
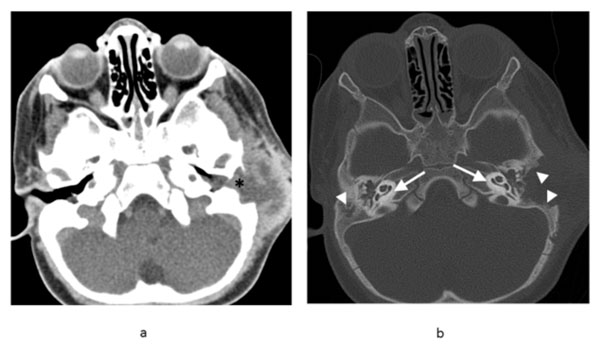
Axial contrast enhanced CT soft tissue window (a) of the head shows soft tissue mass and fluid in the subcutaneous soft tissues surrounding the left ear with soft tissue obliterating the external auditory canal (asterisk). Bone window (b) shows temporomastoid bone destruction bilaterally (arrowheads), greater on the left, with relative sparing of the bony labyrinths (arrows).
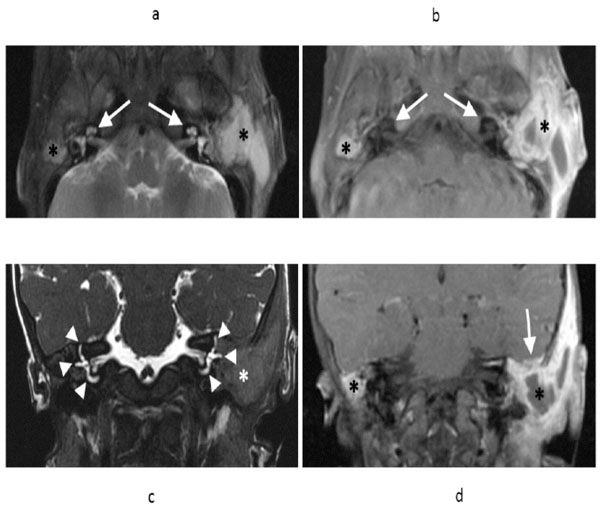
Axial T2 weighted fast spin echo (FSE) MR image (a) and axial T1 weighted FSE post-gadolinium T1 (b) demonstrates fluid and enhancing soft tissue filling the mastoid air cells bilaterally (asterisks), greater on the left. Note preservation of the bony labyrinths bilaterally (arrows). Coronal T2-weighted sequence (c) demonstrates preservation of the semicircular canals bilaterally (arrowheads), despite adjacent soft tissue abnormality (asterisk). Coronal T1 weighted FSE contrast enhanced T1 (d) demonstrates the fluid and enhancing soft tissue involving the mastoids bilaterally (asterisks). On the left, there is extension along the tegmen tympani with minimal adjacent dural thickening and enhancement without adjacent brain parenchymal involvement (arrow).
Definitive diagnosis was confirmed with biopsy. Pathology revealed positive immunohistochemical staining for CD1a, which is pathognomonic for LCH. Skeletal survey, CT of the chest, abdomen, and pelvis, and bone marrow biopsy were all negative for signs of further involvement. Audiometry demonstrated left-sided conductive hearing loss.
Upon diagnosis, the patient proceeded with standard induction therapy including vinblastine and prednisone for six weeks followed by repeat imaging. She had an incomplete response, and thus received a second induction. She then went on to receive continuation therapy with vinblastine and prednisone. Interim imaging with CT (Fig. 4) and MRI (Fig. 5) has suggested a positive response to therapy with no progression of disease. Additionally, her hearing improved on subsequent audiometry.
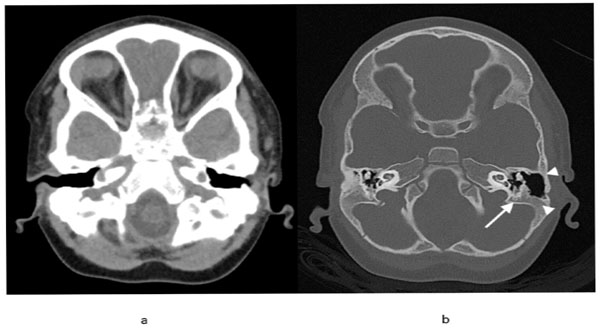
Follow-up axial nonenhanced soft tissue window CT (a) of the head showing complete resolution of the soft tissue thickening and fluid collection in the left temporal bone region. Axial bone window CT (b) shows interval remineralization of the mastoid bone (arrowheads), including the area surrounding the semicircular canals and facial nerve canal (arrow). On the right, the fluid in the mastoid air cells has resolved and interval mineralization is noted.
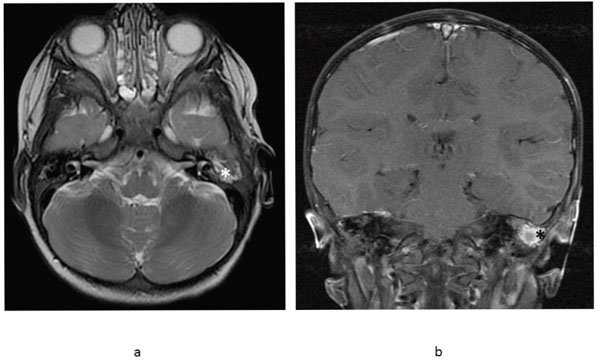
Follow-up axial T2 weighted FSE MR image (a) demonstrates interval decrease in the size of the left tympanomastoid mass (asterisk) consistent with improving Langerhans' cell histiocytosis. Coronal T1 weighted FSE contrast enhanced T1 (b) demonstrates the fluid and enhancing soft tissue involving the left mastoid has improved (asterisk) and has resolved on the right.
DISCUSSION
Although the exact etiology and pathogenesis are uncertain, LCH is described as the clonal proliferation of Langerhans cells. Many etiologic theories exist, primarily neoplastic versus inflammatory, with many possible predisposing factors identified including genetic, immunologic, and viral [1, 3]. Regardless of the etiology, LCH has an unpredictable natural history. Due to its rarity and widely varied clinical presentation ranging from single system to multisystem involvement, LCH is commonly missed.
For instance, clinical manifestations of temporal bone LCH can be similar to more common conditions such as mastoiditis, recurrent or chronic otitis media, and otitis externa, requiring a very high index of suspicion for early detection. In a study by Saliba et al., the most common presentation of temporal bone LCH was a temporal bone mass (70%) and otitis media and externa (60%) [1]. Temporal bone lesions can also be associated with headaches, otalgia, and conductive hearing loss. Based on audiometry, our patient demonstrated conductive hearing loss. Of note, the bony labyrinth is generally spared in LCH, protecting the cochlea and vestibular apparatus and preserving sensorineural hearing, as was the case in our patient [1]. This finding is also a radiologic clue to the diagnosis of LCH.
Head and neck LCH may compress or invade neural structures, mainly the optic, facial, oculomotor, and trigeminal nerves. Other signs and symptoms of head and neck LCH include scalp vesicles, facial rashes, loose teeth, periodontitis, and exophthalmia [1, 4]. Therefore, a thorough neurologic and skin examination should be performed on all children with suspected LCH. Temporal LCH can also present with extracranial manifestations due to the fact that LCH affects reticuloendothelial cells throughout the body. For example, skin, liver, spleen, lung, lymph node, pituitary gland, and bone marrow can be involved in addition to bone [5]. Due to involvement of the skull bones, this patient was classified as “central nervous system (CNS) risk” according to recent diagnostic guidelines, meaning that she may be predisposed to diabetes insipidus and other potentially irreversible CNS manifestations [7].
Due to the profound rarity and clinical mimicry of other more common diseases, temporal bone LCH may be most accurately detected by radiologic studies. Many different imaging modalities are used in the identification of LCH including plain film radiography, CT, and MRI. On a radiograph, LCH typically appears as lytic or “punched out” lesion. Once LCH is suspected, a full skeletal survey or bone scan should be performed for further staging. The gold standard for identification and monitoring of bony LCH is CT [1, 4, 5]. A CT scan demonstrates well-demarcated radiolucent areas and can identify bony destruction of the petrous apex better than MRI [8]. However, MRI is more useful in delineating soft tissue and intracranial extension, and thus was used in our patient. On T1-weighted imaging, LCH involvement is characterized by a hypointense signal, and on T2-weighted imaging, it demonstrates a hyperintense signal. Additionally, LCH lesions show a marked enhancement with gadolinium (Fig. 3) [4]. Positron emission tomography has recently been found useful in identifying extraskeletal LCH and evaluating response to treatment, however further studies need to be completed before routine clinical use can be recommended [3].
A high clinical index of suspicion for LCH should be raised when lytic lesions of the temporal bone are found on imaging, especially if they demonstrate sparing of the bony labyrinth. Although rare, the bilaterality of temporal bone lesions may be highly suggestive of LCH since few other disease processes have this capability in children. The diagnosis should be confirmed by immunohisto-chemical staining of S-100 protein and/or CD1 antigen [1]. Once a diagnosis is made, most will go on to receive chemotherapy with prednisone and vinblastine. The prognosis of LCH localized to the temporal bone is favorable; however multisystemic involvement portends a worse survival rate. With earlier identification of LCH by clinical and radiologic clues, survival rates will likely improve.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
ACKNOWLEDGEMENTS
Declared none.

